Tutorial
Tutorial
Database Introduction
Our research group established a Carbapenem-resistant Enterobacteriaceae (CRE) network to investigate the epidemiology of CRE in China starting from 2014.
There were 2 stages to this study: first, from 2012 to 2013, we collected 150 CRE isolates from 16 tertiary hospitals, and second, starting in 2014, we expanded the collection area.
The number of participating hospitals increased from 25 in 2014 to 65 in 2016.
Peking University People’s Hospital was the lead unit in this project and was responsible for the collection, identification, and sorting of isolates.
We downloaded 846 Klebsiella pneumoniae isolates form refseq database, did the kleborate type, and incorporated them into this database
After comparing with the strains in this database, we will output the virulence genes and antimicrobial genes carried by the strains closest to the isolate. However, since the isolate itself may carry different virulence or antimicrobial genes from the strains in the database, for example, a antimicrobial plasmid is obtained.
For the sake of insurance, this
database will also compare the submitted data with the VFDB database and the resfinder database, and output the virulence
genes and antimicrobial genes of the isolate sequence for comparison.
If it is a nosocomial outbreak, the evolution of the strain
can be briefly viewed from this.
Webserver Introduction
The purpose of PathoTracker is to solve how to find the isolate closest to the newly sequencing data
and timely detect the outbreak of the isolates in the large database.
If the sequencing samples to be detected in the same area over a period of time, by comparing with the
PathoTracker, the results of the output neighboring isolates are similar (here, the closest isolates
are basically the same with sample), the output virulence genes and antibiotic resistance genes are
basically the same, and it can be considered that there may have an isolate outbreak.
At the same time, this service can also trace the source of samples and the geographical and phylogenetic
evolution. In particular, PathoTracker incorporates not only isolates from China but also isolates submitted
worldwide in public databases, at which point its possible to trace the evolution of isolates in both
spatial as well as temporal dimensions.
PathoTracker relies on CRE network. For all the self-collected isolates, we measured the minimum inhibitory
concentration (MIC) of the 23 antimicrobials by utilizing broth microdilution method.
The 23 antimicrobials were as follows:
meropenem (MEM), imipenem (IMP), ertapenem (ETP), cefoxitin (FOX), cefepime (FEP),
ceftazidime (CAZ) ,ceftazidime/avibactam (CAZ‑AVI), ceftazidime/clavulanic acid (CLC), cefotaxime (CTX),
ceftriaxone (CRO), Cefoperazone/sulbactam (CSL), Piperacillin/tazobactam (TZP), amikacin (AMK), ciprofloxacin (CIP),
levofloxacin (LVX), meromycin (MIN), colistin (COL), tigecycline (TGC), chloramphenicol (CHL),
fosfomycin (FOS), aztreonam (ATM), aztreonam/avibactam (ATM-AVI), cefotaxime/clavulanic acid (CTC)
The advantage of this service is the ability to report genomic and susceptibility information for isolates close to the sample. Such that the clinic is capable for dramatically reducing the time required for pure culture, give patients timely precise medication, symptomatic treatment, rescue patients at risk, and restrain abuse of antibiotics, based on genomic information and self-collected susceptibility information from PathoTracker service report.
Example file format and email address
For preassembled partial, complete genomes, the input sequence must be in FASTA format.
The allowed alphabet (not case sensitive) is the following:
For DNA sequences: A C G T and N (unknown)
PathoTracker is only available for Klebsiella pneumoniae right now. More pathogens will be added..
Click Example sequence format to download sample file and view the format
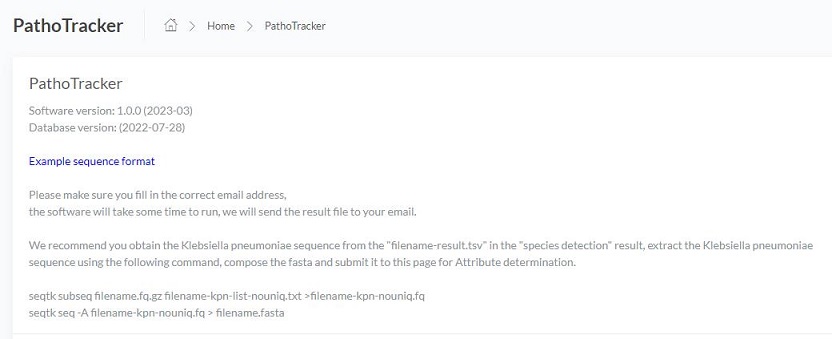
Please make sure you fill in the correct email address,
the software will take some time to run, we will send the result file to your email.
Limit parameters for comparsion
All of the following parameters are the limits you wish to set when comparing with public database data
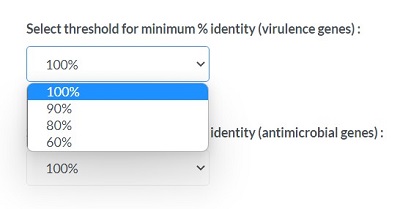
You can choose threshold for minimum % identity (virulence genes)
You can choose threshold for minimum % coverage (virulence genes)
You can choose threshold for minimum % identity (antimicrobial genes)
You can choose threshold for minimum % coverage (antimicrobial genes)
For each limitation, You can choose 100% 90% 80% 60% level

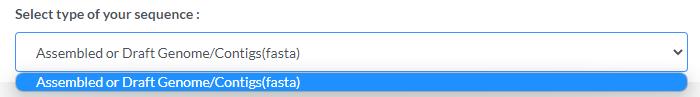
Submit data type
Please select the type of data to submit
For metagenomic data type:
fasta format is support
Only 1 fasta file can be uploaded
For unassembled TGS sequencing reads
We recommend use "seqtk seq -A filename-kpn-nouniq.fq > filename.fasta" to get fasta
Only 1 fasta file can be uploaded
If "Culture isolate (NGS)" is chosen:
Only 1 assembled fasta file can be uploaded
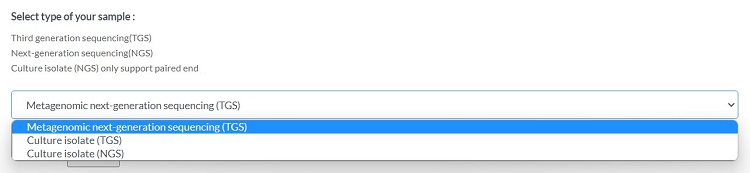
For mNGS raw data (fastq), we will provide clean the data, remove its human reads in the future.
For NGS raw data (fastq), we will provide clean the data, sequence assembly in the future.
We strongly recommend that for Metagenomic next-generation sequencing (TGS) data, it is better to have the reads of
Klebsiella pneumoniae greater than about 1000. The results report will show how many reads Klebsiella pneumoniae has.
The more reads, the more accurate the results will be.
Result
The results of the PathoTracker will be sent to you by email.
Since it takes time to generate the result,
please make sure you fill in your email address correctly so that we can send you the results.
The results of the PathoTracker will also be displayed on the results page.
The reads matching results will be sent to your email
The result of PathoTracker
The result of sequences comparing with resfinder database and VFDB database
Submission
The data type should be:
Metagenomic next-generation sequencing Third generation sequencing(TGS) remove host background
Click Example sequence format to download sample file and view the format
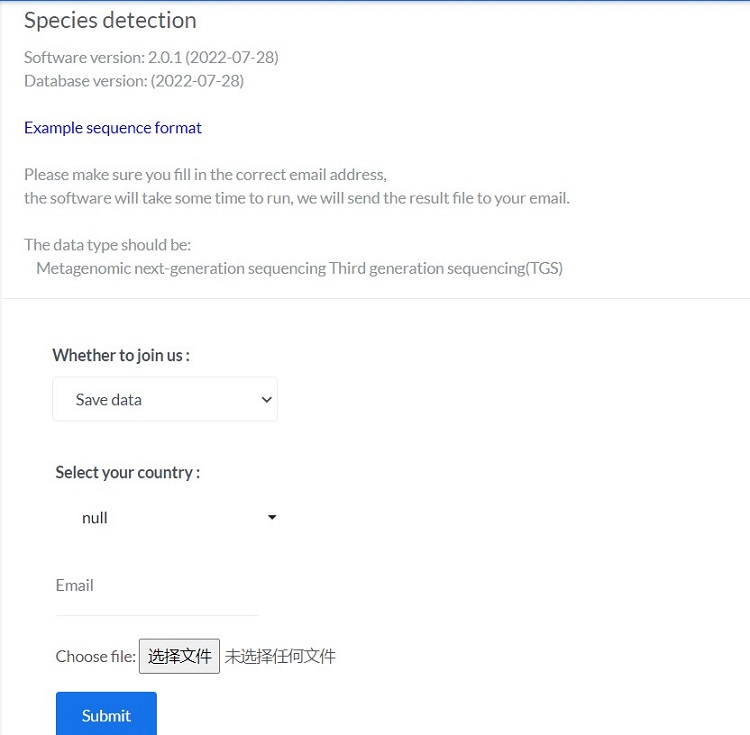
Please make sure you fill in the correct email address,
the software will take some time to run, we will send the result file to your email.
Result
The results of the PathoTracker will be sent to you by email.
Since it takes time to generate the result,
please make sure you fill in your email address correctly so that we can send you the results.
The results of the PathoTracker will also be displayed on the results page.
We clean the data, remove its human origin and detect species
The reads matching results will be sent to your email
Webserver Introduction
Phylogenetic tree enables uploading assembled sequencing data,
constructing the core genome of these sequences through Roary ,
and building phylogenetic tree of the core genome through iqtree2 .
Isolate sequences will be annotated using Prokka
Roary will be used to calculate the core genome for phylogenetic analysis, which takes annotated assemblies in GFF3 format (produced by Prokka)
Iqtree2 will be used to infer phylogenetic trees by maximum likelihood in core genome alignment
Since the isolate genome is large magnitude, so it takes a long time to obtain the genome sequence for constructing the phylogenetic tree by conventional mapping and find SNP or MEGA, PathoTracker uses Roary to extract the core genome.
Example file format and email address
For preassembled partial, complete genomes, the input sequence must be in FASTA format.
The allowed alphabet (not case sensitive) is the following:
For DNA sequences: A C G T and N (unknown)
Click Example sequence format to download sample file and view the format
Please make sure you fill in the correct email address, the software will take some time to run, we will send the result file to your email.
Tree number
Phylogenetic trees require a minimum of 4 sequences,
and this webserver currently can handle a maximum of 6 sequences
Submit data type
Please select all fasta files at the same time before uploading them
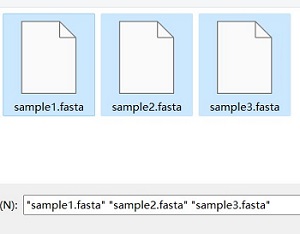
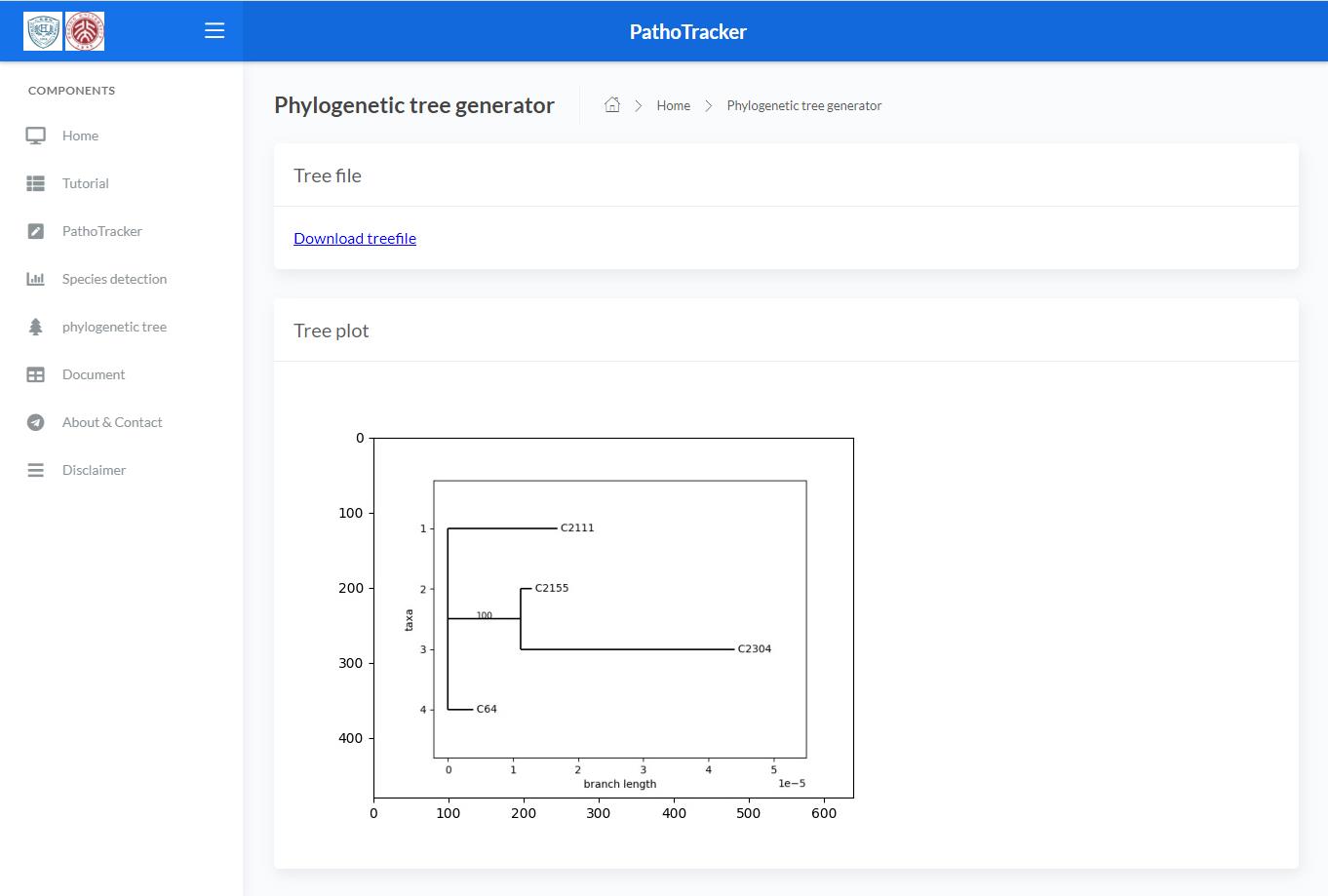
Tree result
The results of the phylogenetic tree construction will be sent to you by email.
Since it takes time to generate the phylogenetic tree,
please make sure you fill in your email address correctly so that we can send you the results.
The results of the phylogenetic tree will also be displayed on the results page.
Statement
We welcome you to join the CRE network. If you do not want to keep your data in the database after using this database, please tick "Do not keep data" when submitting the data, and we will delete the data after the data analysis is completed.
If you would like to keep your data and join us, please select yes
If you select yes, the city is mandatory
We recommend that you use Chrome, Safari, Firefox, 360 browsers to successfully select your country.
If you would like to join us but do not have one of these browsers, please do not hesitate to contact us by email, thank you.